Biological reactions are regulated by thermodynamically and/or kinetically controlled molecular recognition events that occur among biomolecules. Dr. Ohkanda’s research interests lie at the interface of organic chemistry and biology, particularly in the areas of molecular recognition and its application to structure-based design of synthetic agents and chemical tools that can be used for better understanding important biological processes, such as protein-protein interactions (PPIs). Her approaches include designing multivalent agents that are capable of binding to large and featureless interfaces of PPIs, where small modular compounds consisting of various functional groups are assembled by tethering with linkers, metal chelation, or target-guided chemical ligation. Her principal focus also includes chemical biological study of 14-3-3 PPI inducers, which are antitumor agents derived from diterpene glucosides, fusicoccins. Her team is currently working on identifying their intracellular binding proteins as well as elucidating the mechanism of biological functions. Exploration of synthetic molecules that regulate transient biomolecular interactions implicating intrinsically disordered proteins is also underway in the group.
Developing clinically relevant synthetic agents that are capable of disrupting protein-protein interactions (PPIs) is now a major goal of scientific research. In an effort to explore new methodologies that are applicable to the design of synthetic PPI inhibitors, we study a new strategy based on the assembly of small module compounds to create multivalent mid-sized agents. The approaches include i) covalent tethering to create anchoring enzyme inhibitors, ii) metal-chelating-based ligand assembly, and iii) chemical ligation templated by a targeted protein. These strategies have been shown to be useful for synthesizing minimally sized synthetic agents for disrupting PPIs and may enable development of agents that are applicable to inhibition of intracellular PPIs.
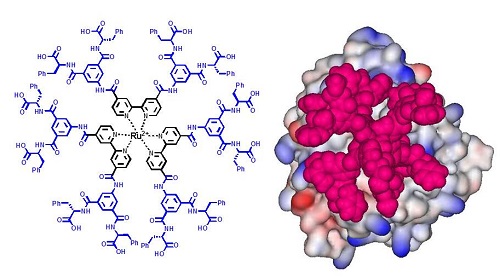
Antibody-like metal complexes featuring large surface areas and multivalency for protein surface recognition are attractive scaffolds for the development of PPI inhibitors as well as for metal-mediated protein assembly. We expected that the formation of metal complexes with ligands containing various functional groups would facilitate introduction of synthetic antibody-like structural diversity into the agents, and that the resulting metal-complex libraries may be useful for identifying lead compounds for various protein targets. To test the feasibility of metal complexes as PPI-directed agents, we have been working on a series of transition metal complexes, including ruthenium tris(bipyridine)s. Our studies have verified that these complexes provide a useful scaffold for PPI agents, demonstrating that the size of the complex, the positioning of the functional groups, and the complementary charge distribution were all found to have a considerable effect on the complex’s affinity for and inhibition of targeted proteins. [Chem. Commun. 2009]
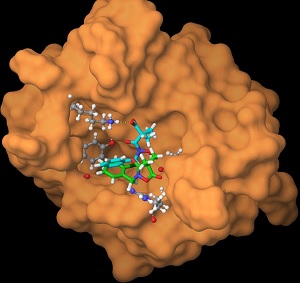
Reducing the molecular weight of a protein-binding agent while retaining its affinity and selectivity for the targeted protein surface is challenging task. One strategy for addressing this problem is to synthesize PPI inhibitors on a targeted protein surface in situ by covalently assembling relatively small module compounds. The resulting agents should bind more readily to the protein surface than either of the module compounds alone due to the effect of additive binding energies. To evaluate the utility of the covalent assembly for in-situ generation of PPI inhibitors, we are investigating ligation reactions guided by 14-3-3 proteins, using fusicoccins and modified peptides or peptidomimetics, to see if we can generate 14-3-3 inhibitors in cells. [J. Am. Chem. Soc. 2015, Mol. BioSyst. 2013]
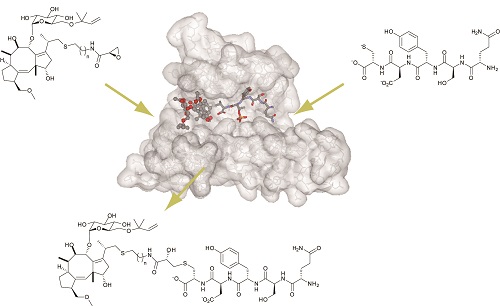
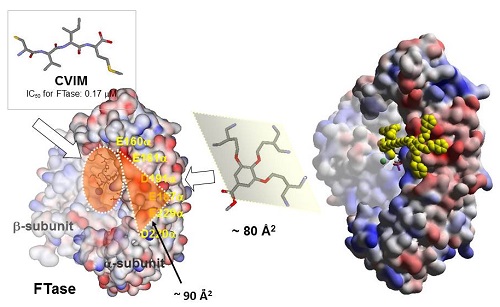
A critical question in developing clinically relevant PPI inhibitors is how to reduce molecular size while retaining the binding affinity and selectivity of the agents. One of our approaches to address this issue is the anchoring strategy in which two modules designed for cavity-binding and for surface-binding are covalently linked on another with an appropriate spacer. The cavity-binding module should bind into the active pocket in a selective manner and anchor the entire molecule near the cavity, thus allowing for a minimally sized surface module to bind to the flat protein surface. We have applied this methodology to two structurally and functionally related metalloenzymes, protein farnesyltransferase (FTase) and type-I gernylgeranyltransferase (GGTase I). These enzymes are implicated in activation of oncogenic Ras in cells, thus being studied for many years as the potential targets of antitumor agents. Our initial project was to design the bivalent inhibitors that disrupt protein-protein interactions between K-Ras and both FTase and GGTase I. Our results demonstrated that the series of bivalent compounds not only show remarkable inhibition efficacy for inhibiting K-Ras prenylation, but also exhibit dual inhibition activity for both FTase and GGTase-I recognizing the common surface structure. We are currently working on expanding this approach to other clinically relevant PPIs. [Bioorg. Med. Chem. 2013, J. Am. Chem. Soc. 2011]
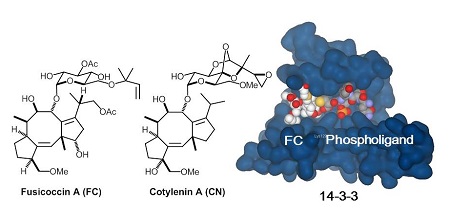
Structurally related diterpene glucosides, fusicoccin A (FC-A) and cotylenin (CN), exhibits distinct biological activities. While FC-A is phytotoxic, CN induces differentiation of human leukemia cells and significantly suppresses tumor growth in animal models. These natural products have been shown to stabilize interactions of 14-3-3 proteins and phosphorylated ligands in vitro. We are interested in addressing fundamental questions, 1) what is th e mechanism of action of antitumor activity of the natural products and related synthetic analogs, 2) why do FC-A and CN show distinct biological activity despite of their structural similarity, and 3) can we design chemical tools for exploring 14-3-3 PPI network in cells. By a collaboration with Prof. Kato’ s research group, we are working on chemical biological study using FC-based agents to identify intracellular binding proteins as well as to elucidate their mode of action. The techniques used in this project include synthetic organic chemistry, biochemistry, molecular biology, and cell biology. [ACS Chem. Biol. 2013, Chem. Biol. 2013, Angew. Chem. Int. Ed. 2012]
|
Loading player ...
|
Isoprenoids are essential for survival in all organisms. Animals synthesize isoprenoids from mevalonic acid, whereas most pathogenic bacteria and the malaria parasites utilize a distinct pathway, the nonmevalonate (methylerythritol phosphate; MEP) pathway. Specific inhibitors of the MEP pathway therefore hold promise as a new class of antibiotic, antituberculosis, and antimalarial drugs. We are focusing on the first enzyme in the MEP pathway, 1-deoxy-D-xylulose-5-phosphate synthase (DXS), and working on developing small molecule-based agents that specifically inhibit DXS, resulting in antibacterial and antimalarial activities. [Chem. Commun. 2013]